We develop a wide variety of statistical and data science methods and workflows for biological (microbial) data across different biological time and spatial scales.
.
Compositional changes of cell types are main drivers of biological processes.
However, their detection through single-cell experiments is difficult due to the compositionality of the data and
low sample sizes.
scCODA, a Bayesian model addressing these issues, enables the study of complex cell type effects in disease,
and other stimuli. scCODA demonstrates excellent detection performance and could identify experimentally verified
cell type changes that were missed in original analyses.
The method was developed by Johannes in collaboration with the groups of
Benjamin Schubert and
Fabian Theis.
You can read more about scCODA in Nature Communications and readthedocs.
.
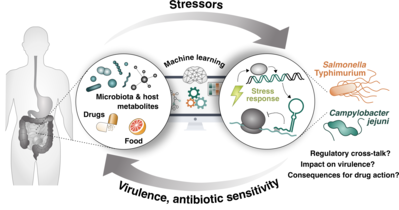
In this project, we aim to identify stressors and bacterial regulatory pathways that control host adaptation and antibiotic sensitivity. By using a high-throughput chemical genomics approach, we will explore chemical stimuli from the host environment that induce stress response pathways in Salmonella and Campylobacter, and elucidate the underlying molecular crosstalk between sensory pathways of these microbes. This project is part of the bavarian Bayresq research consortium, and is done in close colaboration with members of Dr. Cynthia Sharma's and Dr. Ana Rita Brochado's labs. For more info on the role of our group in this project you can contact Roberto
.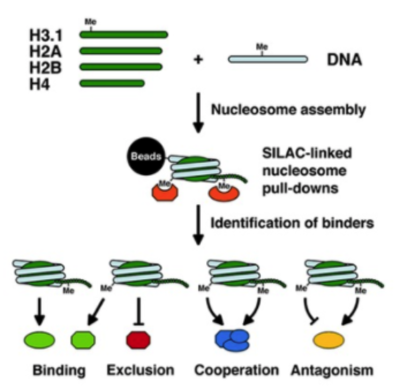
In this project, we combine chemical biology, biochemical, and proteomic approaches with cell biology and computational analyses to unravel how epigenetic effector molecules can read DNA and histone modification patterns and how they recognise different chromatin modification states. Our goal is to decipher the “epigenetic code” by identifying epigenetic reader molecules that can integrate information from multiple chromatin modifications and to understand how these factors operate at the molecular level both in healthy and pathological conditions.
This project is done in close collaboration with Dr. Till Bartke from the Institute of Functional Epigenetics at Helmholtz Zentrum München. In our group Mara is responsible for this project.
.
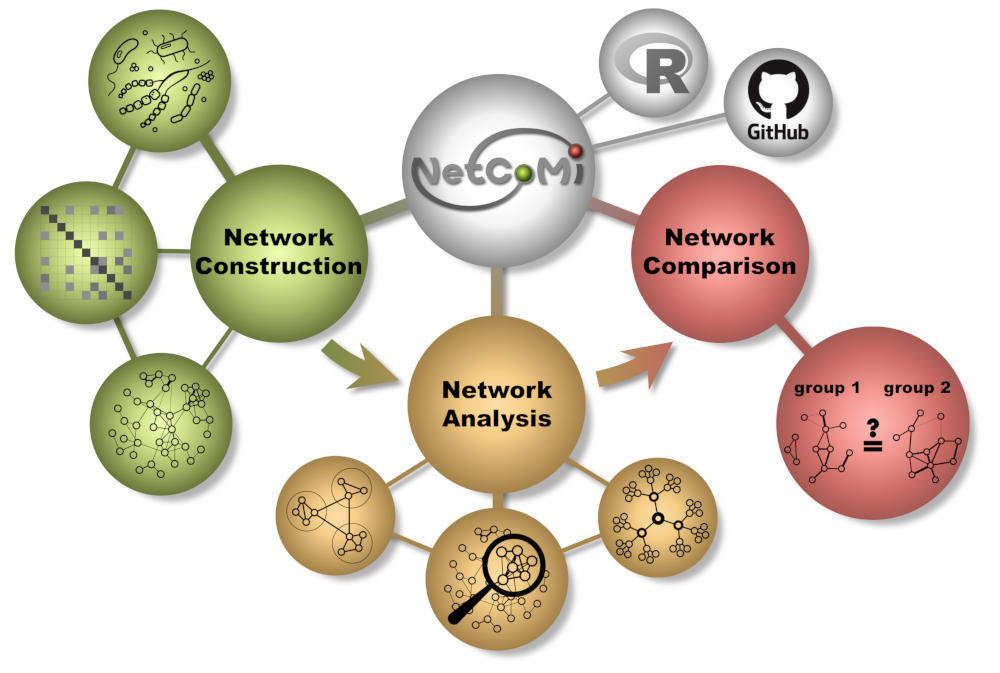
NetCoMi is an R package that provides an extensive toolbox for constructing, analyzing, and comparing networks for microbial compositional data. It also offers the ability to quantify network differences and thus enables insights into whether single taxa, groups of taxa, or the overall network structure change between groups. The package was developed by Stefanie as part of her Ph.D. thesis, in collaboration with her supervisors and the group of Prof.Dr. Erika von Mutius at Helmholtz Zentrum München. The manuscript describing the workflow and theory behind the package is published in Briefings in Bioinformatics.
.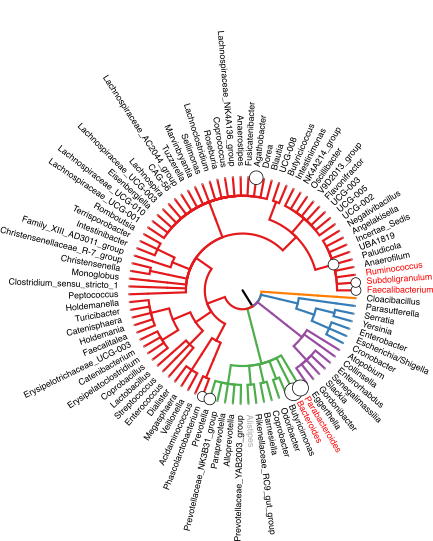
Microbial or cell type compositions from high-throughput sequencing experiments are often ordered by a tree structure, such as taxonomic or cell lineage trees.
The tascCODA method, which was developed by Johannes,
utilizes this structure to dynamically infer compositional changes on subgroups of the composition.
Compared with other methods, tascCODA has very good detection accuracy and gives a more parsimonious picture of the compositional shifts in the data.
The method was published in Frontiers in Genetics, and the Python implementation can be found on our github page.
.